



")
Com o desenvolvimento de novas técnicas de sequenciamento como o Sequenciamento de Nova Geração, o volume de variantes observado aumentou drasticamente. Neste sentido, o uso de uma nomenclatura de variantes padronizado para identificar e gerenciar a quantidade de dados de sequência de variantes se tornou necessário.
O que é uma variante genética?
Variante é uma alteração na sequência de nucleotídeos em uma posição específica do genoma em relação a um genoma de referência.
Variantes podem ocorrer em qualquer posição do genoma, incluindo as regiões gênicas, ou seja, regiões que carregam a informação para síntese de proteínas. Estas regiões são as de maior interesse em pesquisas clínicas na busca por variantes relacionadas com determinados distúrbios.
O impacto de uma alteração em uma região de codificação de um gene, depende principalmente de como essa variante afeta a síntese da proteína depois da tradução.
Bancos de variantes
A complexidade e variações das sequências do genoma humano e de outros organismos exige excelentes recursos de bioinformática para garantir a organização do gerenciamento de dados. Os dados genômicos obtidos em testes clínicos e laboratoriais são utilizados para o melhor manejo de pacientes e outros estudos epidemiológicos. Por isso, é de extrema importância manter uma comunicação precisa e consistente dentro da comunidade cientifica.
Para que o acesso a informação sobre novas variantes seja disponível para todos, existem bancos de dados online ou seja, repositório de acesso normalmente aberto, que agrupam estas sequências. Diferentes bancos de dados de variantes genéticas foram desenvolvidos e são divididos, por exemplo, em banco de variantes específicas de uma doença ou de dados populacionais.
Estes bancos podem ser abastecidos por pesquisadores do mundo todo e os dados de variantes depositados passam por análise antes de serem aceitos para divulgação. Estas informações são especialmente importantes para realizar testes de diagnóstico e avaliação de risco genético.
Assim como através do Projeto Genoma Humano, padrões foram estabelecidos para uma documentação uniforme e agrupada de dados de sequências, a nomenclatura de sequências variantes também deve seguir algumas regras.
Como é a nomenclatura de variantes genéticas?
Como citamos anteriormente, houve um grande aumento do volume de variantes identificadas em diferentes organismos, principalmente nos últimos anos com o desenvolvimento de novas tecnologias de sequenciamento.
Para evitar confusão quanto ao significado da variante, foi desenvolvido um padrão para essa linguagem, o chamado padrão de nomenclatura de variantes HGVS (Human Genome Variation Society). O padrão é usado em todo o mundo, especialmente em saúde humana e diagnósticos clínicos.
Todas as variantes descritas devem conter a clara indicação se a mudança foi determinada experimentalmente ou deduzida teoricamente. Além disso, todas as variantes devem ser reportadas baseadas em um genoma de referência público e reconhecido, como as do NCBI. Entenda melhor sobre genomas de referência aqui: O que é um genoma de referência?
- Para indicar qual referência foi utilizada, são utilizadas nomenclaturas como:
LRG_# Locus Reference Genomic (exemplo LRG_199, LRG_199t1)
NC_# Genoma completo (Exemplo: NC_000023.10)
NG_# Genoma incompleto Exemplo: NG_013231.1
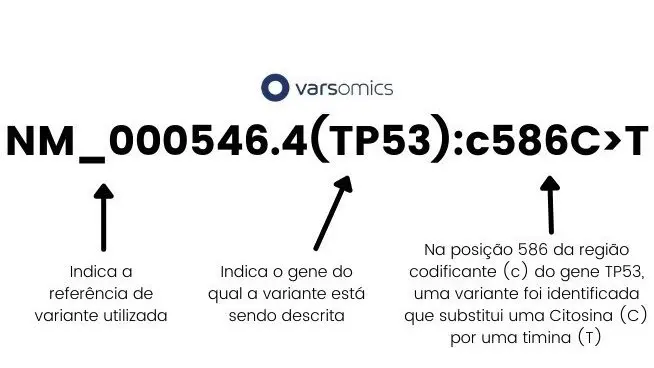
NM_# mRNA Exemplo: NM_003506.2
NR_# ncRNA- RNA não codificante Exemplo: NR_002293.1
NP_# Proteína. Exemplo: NP_003987.1
- Para indicar o tipo de sequência de referência usada, os seguintes prefixos são utilizados:
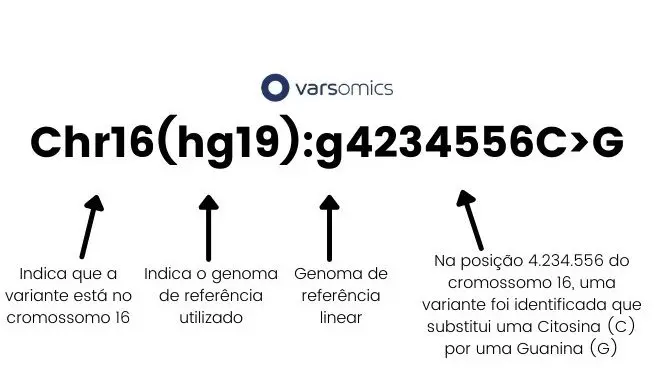
g. para uma sequência de referência genômica linear
m. para uma sequência de DNA mitocondrial
c. para uma sequência de DNA codificadora (gene)
n. para uma sequência de DNA não codificante
o. para uma sequência de referência genômica circular
p. para uma sequência de proteína
r. para uma sequência de referência de RNA (transcrição)
- Variantes podem ser descritas baseado na sua posição no genoma ou em um gene específico. As descrições em nível de DNA, RNA e proteína devem ser:
A nível de DNA – 123456A>T: número referente a posição do nucleotídeo afetado, nucleotídeos em MAIÚSCULAS.
A nível de RNA – 76a>u: número referente a posição do nucleotídeo afetado, nucleotídeos em letras minúsculas.
A nível de proteína– Lys76Asn: Indica o(s) aminoácido(s) afetado(s) usando código de três ou uma letra seguido por um número símbolos de aminoácidos atribuídos.
- Algumas abreviações utilizadas para descrever a alteração causada na variante:
“>” (maior que) Utilizado para indicar substituição. Exemplo: g.12345A>T substituição de um A por T.
“dup” indica uma duplicação; exemplo c.49dupA
“ins” indica uma inserção; exemplo c.39_40insG
“inv” indica uma inversão; Exemplo c.76_83inv
“fs” indica um frameshift; Exemplo p.Arg456GlyfsTer17
“cen” indica o centrômero de um cromossomo
“chr” indica o cromossomo onde a variante está localizada. Exemplo: chr12:g.34232311C>T
“pter” indica o primeiro nucleotídeo de um cromossomo
“qter” indica o último nucleotídeo de um cromossomo
“sup” indica um cromossomos supranumerários (sSMC) – pequenos cromossomos anormais que não podem ser identificados de forma inequívoca por técnicas clássicas de citogenética.
“gom” indica ganho de metilação
“lom” indica perda de metilação
“met” indica uma metilação
Exemplos a nível de DNA:

Exemplo a nível de proteína:

- Alguns caracteres também são utilizados para nomear variantes:
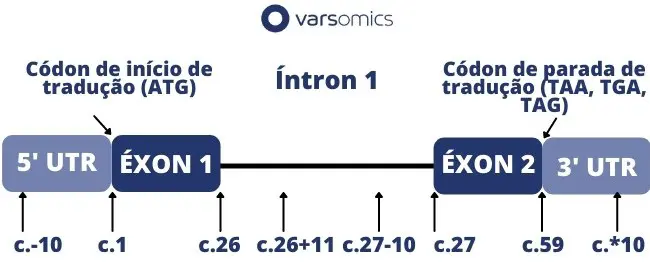
“+” (sinal de mais) usado para numerar a posição de um nucleotídeo em relação a outro. nucleotídeos na extremidade 5 ‘de um íntron são numerados em relação ao último nucleotídeo do exon diretamente a montante, seguido por um “+” (mais) e sua posição no íntron. Exemplo: c.123+45A>G
“-” (sinal de menos) nucleotídeos na extremidade 3 ‘de um íntron são numerados em relação ao primeiro nucleotídeo do exon diretamente a jusante, seguido por um “-” e sua posição fora do intron. Exemplo: c.124-56C>T
“?” (interrogação) indica incerteza na posição do nucleotídeo ou aminoácido g.(?_234567)_(345678_?)del
“( )” (parênteses) são usados para indicar incertezas e predições; NC_000023.9:g.(123456_234567)_(345678_456789)del, p.(Ser123Arg)
“*” (asterisco) indica um códon de terminação de tradução. Exemplo c.*43C>T ou p.Trp41*
“_” (sublinhado) indica um intervalo entre posições. Exemplo: g.12345_12678del
“[ ]” (colchetes) indica alelos, que inclui múltiplas sequências inseridas em uma posição e inserções de uma segunda sequência de referência.
“;” (ponto e vírgula) Utilizada para separar variantes e alelos. Exemplo: g.[456434A>G;542324G>C] ou g.[456434A>G];[ 542324G>C]
“,” (vírgula) utilizada para separar diferentes transcritos/proteínas sintetizados a partir de um alelo. Exemplo: g.[432a>u, 211_253del]
“:” (dois pontos) usado para separar a sequencia de referência da variante que está sendo descrita. Exemplo: NC_000011.9:g.12345611G>A
“?” (interrogação) indica incerteza na posição do nucleotídeo ou aminoácido g.(?_234567)_(345678_?)del
“^” (acento circunflexo) indica “OU” em casos de incerteza; c.(370A>C^372C>T)
“=” (igual) indica que uma sequência foi testada, mas não foi alterada. Exemplo: p.(Arg234=)
“/” (barra pra frente) indica mosaicismo.
“//” (dupla barra para frente) indica quimerismo.
“|” (barra vertical) indica que não houve uma alteração na sequência, porém ocorreu uma modificação, como por exemplo uma metilação.
Alguns exemplos de nomenclatura de variantes
Variante descrita de acordo com sua posição no genoma:
Variante descrita de acordo com sua posição em um gene:
Para consultar o documento completo com todas as informações sobre como nomear uma variante acesse: Sequence Variant Nomenclature – HGVS
Referência:
den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat. 2016;37(6):564-569. doi:10.1002/humu.22981
![]()