

")
Mutations in the Human Immunodeficiency Virus Related to Antiretroviral Protease Inhibitors: Literature Review
Juliano Soares de Paula 1, Allyne Cristina Grando1
1Universidade Luterana do Brasil (ULBRA). Canoas, RS, Brasil

Resumo: O vírus da imunodeficiência humana (HIV) é conhecido como causador da síndrome da imunodeficiência adquirida (AIDS), uma importante doença que se espalha ao mundo há 40 anos sem apresentar uma cura definitiva. Em meados da década de 80 surgiu o primeiro fármaco com efeito de retardar o avanço viral no organismo, junto a isto surge também as mutações do HIV frente a pressão farmacológica que gera um mecanismo de adaptação para seguir seu processo replicativo. Uma década após surgir o primeiro antirretroviral (ARV) é aprovado uma nova geração de fármacos focados na fase final do ciclo vital do vírus, conhecido como inibidor de protease IP. O objetivo deste trabalho foi realizar uma revisão da literatura sobre a resistência aos antirretrovirais da classe dos inibidores da protease utilizados no tratamento do HIV relacionando-as com mutações específicas do vírus.
Palavras-chave: HIV, mutações e inibidores de protease.
Abstract: The human immunodeficiency virus (HIV) is known to cause acquired immunodeficiency syndrome (AIDS), an important disease that has spread to the world for 40 years without showing a definitive cure. In the mid-1980s, the first drug with the effect of delaying viral progress in the body emerged, along with this also appears HIV mutations in the face of pharmacological pressure that generates an adaptation mechanism to follow its replicative process. A decade after the first antiretroviral (ARV) appeared, a new generation of drugs focused on the final stage of the virus’s life cycle, known as IP protease inhibitor, is approved. The objective of this work was to carry out a literature review on the resistance to antiretroviral drugs of the class of protease inhibitors used in the treatment of HIV, relating them to specific mutations of the virus.
Key-words: HIV, mutations and protease inhibitors.
Introdução
O vírus da imunodeficiência humana (HIV), é o causador de uma grave e importante pandemia a década de 1980, quando surgiu o primeiro caso nos Estados Unidos (1). Estatísticas globais relatam que o ano de 2018 terminou com 37,9 milhões de pessoas infectadas, sendo que aproximadamente 23,3 milhões tem o acesso a terapia antirretroviral (TARV). Desde sua descoberta, doenças relacionadas a Síndrome da Imunodeficiência Adquirida (AIDS) mataram cerca de 32 milhões de pessoas (2). A região mais afetada é a África subsaariana que representa aproximadamente 69% de afetados na população mundial (1). No Brasil, nos últimos anos, tem-se registrado, uma média anual de 39 mil novos casos, totalizando desde 1980 a junho de 2019 uma estimativa de aproximadamente 966 mil pessoas infectadas (3).
A TARV tornou a AIDS uma doença crônica, mas sua eficiência depende da adesão ao tratamento, que tem como objetivo tornar os níveis virais indetectáveis para controle da doença, devido ainda não existir cura. No ano de 1986, nos Estados Unidos, foi aprovado a Zidovudina para tratamento da AIDS (4), sendo no Brasil distribuído somente em 1991 devido a conflitos normativos internacionais que interferem na política brasileira sobre a distribuição dos antirretrovirais (ARV). Em 1995 foi aprovado o primeiro inibidor de protease (IP) o Saquinavir, que chegou ao Brasil no ano seguinte junto ao segundo IP Ritonavir (5,6).
Em 1993 e 1994 deu-se início a estudos sobre combinações dupla ou tripla da TARV, que se tornou padrão mundial em 1996, a qual ficou conhecida como Terapia Antirretroviral Altamente Ativa, conhecido em inglês, como HAART (highly active antirretroviral therapy), sendo composta de três antirretrovirais de duas classes diferentes (4). Hoje, no Brasil, são fornecidos 22 antirretrovirais (ARV) pelo Sistema Único de Saúde (SUS), incluindo a formulação em dose fixa combinada (DFC), que simplificou a TARV em um comprimido ao dia (7). O uso de combinações de fármacos é utilizado para retardar o surgimento de variantes resistentes do HIV, e por sua vez é mais eficaz do que regimes monoterápicos. Porém a utilização dessa combinação de fármacos ser muito empregada nos tratamentos sempre haverá o risco de o vírus desenvolver resistência às drogas (1).
A replicação viral ocorre em alta taxa, sendo em média 10 bilhões de partículas por dia, com uma grande propensão ao erro da transcriptase reversa, associando assim novas mutações ao genoma do vírus, contribuindo para a resistência aos antirretrovirais (8). A resistência aos IPs decorre das mutações no gene que codifica a protease, levando a uma alteração na estrutura proteica da enzima que, como consequência, leva a diminuição no tempo de ligação dos IPs a protease (9). O vírus apresenta mecanismos de adaptação frente ao sistema imune e ao tratamento farmacológico permitindo que o TARV venha falhar após uso prolongado (8). Em consequência o aumento de concentração dos ARV no organismo podem potencializar à indução de citoxicidade e genotoxicidade, apresentando alguns danos como a lipodistrofia, hepatoxicidade, anemia, dislipedemias, entre outros, portanto os riscos de uma exposição duradoura aos ARV podem gerar uma instabilidade genômica, lesionando o ácido desoxirribonucleico (DNA) e os cromossomos, resultado da incorporação dos fármacos ao DNA humano (10).
Portanto, este trabalho teve como objetivo realizar uma revisão da literatura sobre a resistência aos antirretrovirais da classe dos inibidores da protease utilizados no tratamento do HIV relacionando-as com mutações específicas do vírus.
Metodologia
Este trabalho foi realizado através de uma revisão bibliográfica da literatura científica, por meio das plataformas eletrônicas tais como: Google Acadêmico, Scientific Electronic Library Online (Scielo), Biblioteca Virtual em Saúde (BVS), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), ScienceDirect e PubMed nos idiomas português e inglês, com assuntos referentes que contemplassem o objetivo deste trabalho dos últimos 20 anos, onde foram selecionados 25 artigos que enquadraram nesses critérios, com os seguintes descritores: VIH, antirretrovirais, mutações, resistência, inibidores de protease, SIDA, HIV, antiretrovirals, mutations, resistance, protease inhibitors, AIDS. Foram excluídos artigos que foram publicados anterior ao ano 2000, que não fossem nos idiomas português e inglês, e que trouxesse informações incompatíveis com o foco do trabalho.
Vírus da imunodeficiência humana (HIV)
O HIV pertence à classe dos retrovírus e subgrupo dos Lentivírus (que são conhecidos por longos intervalos entre a infecção e os sintomas graves), sua estrutura é de formato esférico com aproximadamente 100 nanômetros de diâmetro, envolvido por um envelope lipoproteico onde estão inseridas as glicoproteínas gp41 que está na membrana celular e o gp120 que é extracelular. Em seu núcleo, dois nucleocapsídeos de capas proteicas formados em sua maioria pelas proteínas P24 e P18 revestem duas cópias idênticas de ácido ribonucleico (RNA), e três enzimas fundamentais (transcriptase
reversa, integrase e protease) (11, 12) conforme ilustrado na Figura 1.
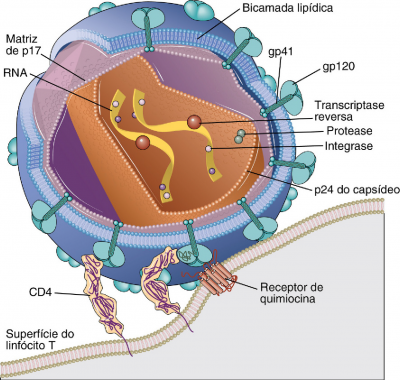
Figura 1: Ilustração do vírus HIV (ABBAS, 2015)
O genoma do HIV mede cerca de 9 kilobytes de comprimento, onde estão inseridos os três genes estruturais: gag (grupo antígeno), pol (polimerase) e env (envelope). Além disso apresenta uma grande variação genotípica devido aos erros de transcrição e replicação viral, dando origem a diversas mutações das sequencias genéticas (11). Existem dois tipos de HIV (HIV-1 e HIV-2); e devido a uma grande variação genotípica o HIV-1 se divide em grupos denominados M (Major), O (Outlier), N (New) e P, que são identificados pela diferença na proteína env, e por sua vez o grupo M é subdivido em A, B, C, D, F, G, H, J e K (11, 12).
Ciclo de vida do vírus da imunodeficiência humana
A entrada do HIV na célula se inicia com a ligação através da alta afinidade da glicoproteína gp120 do vírus, com receptor CD4 do linfócito T e macrófagos; esta interação resulta na exposição da alça V3 da gp120, que se torna apta a ligar-se aos co-receptores CCR5 ou CXCR4, permitindo assim a ativação da glicoproteína gp41, que por sua vez auxilia na conformação das membranas admitindo a fusão do vírus e célula (1,11,13).
Após a fusão o conteúdo do capsídeo é liberado no citoplasma da célula hospedeira, e a enzima transcriptase reversa entra em ação, convertendo o ácido ribonucleico (RNA) viral em uma dupla hélice de DNA viral, onde é transportado ao núcleo celular e integrado ao material genético do hospedeiro através da enzima integrasse (14). Logo após a integração o vírus pode permanecer em estado de latência sendo somente replicado seu material genético mas não transcrito(11). Contudo, quando a célula hospedeira for ativada, os genes virais expressam a transcrição do RNA viral e tradução das proteínas virais, que se movem para a superfície celular formando novos vírus inativos. Em seguida entra em ação a enzima protease que cliva as poliproteínas tornando-as em enzimas estruturais e funcionais, além de ativar o HIV para nova infecção (15), reiniciando o novo ciclo de vida do HIV (Figura 2).
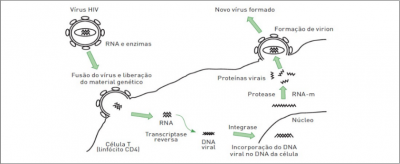
Figura 2: Ciclo de vida do vírus HIV (Cavalheiro, Sprinz, 2013)
Antirretrovirais
O objetivo de uma terapia contra a AIDS é manter indefinidamente a supressão plasmática dos níveis de RNA viral abaixo dos limites de detecção de ensaios sensíveis, com a finalidade de diminuir ou reverter o dano imunológico. Em meados da década de 90, ficou claro que o esquema terapêutico mais adequado para conter a replicação viral teria que ser constituído, pela associação de classes diferentes de ARVs, quando foi introduzido os inibidores de proteases (IPs), que clinicamente, os pacientes apresentaram um aumento da taxa de linfócitos TCD4+ e diminuição da carga viral (16).
Os antirretrovirais com seu aumento de concentração no organismo podem potencializar à indução de citoxicidade e genotoxicidade, onde os inibidores de trancriptase reversa atuam também na inibição da replicação mitocondrial, gerando assim toxicidade que pode apresentar alguns danos como a lipodistrofia e a neuropatia periférica associados a Estavudina; a erupção cutânea e a hepatoxicidade ligados a Nevirapina; e a anemia a Nidovudina (17); assim como outros danos: miopatias, miocardiopatia hipertrófica, Diabetes Mellitus, pancreatite e acidose láctica. Os inibidores de protease podem levar a distúrbios gastrointestinais e dislipedemias (18).
A terapia antirretroviral promove maior expectativa de vida, entretanto está associada ao aumento da adiposidade corporal e cronicidade do estado inflamatório, causando uma série de anormalidades endócrino-metabólicas. Sendo que pacientes em uso de IP há no mínimo 18 meses apresentam duas vezes mais chance de desenvolver lipodistrofia (19).
Mecanismo de ação dos inibidores de protease
A enzima viral protease é um homodímero simétrico composto por dois monômeros de 99 aminoácidos, responsável pelo processamento, ou seja, clivagem das poliproteínas gag e gag – pol, levando a formação de proteínas estruturais e funcionais do vírus HIV, formando uma nova partícula antes do encapsulamento do mesmo. (6, 12, 8, 15) Ele pertence à família das aspartil proteases, contendo dois grupos β-carboxi aspartil (Asp-25 e Asp-25’) no sítio ativo, responsáveis por catalisar a hidrolise das ligações peptídicas (8), conforme ilustrado na Figura 3.
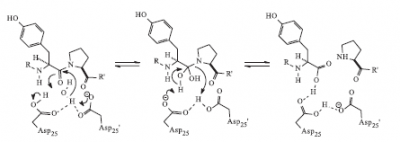
Figura 3: Mecanismo de Hidrolise da protease (Peçanha et al, 2002)
Ambos os resíduos aspartil estão envolvidos na hidrólise, sendo na adição de uma molécula de água à carbonila da amida do substrato, formando um intermediário tetraédrico que apresenta grande afinidade ao sítio catalítico, por onde este intermediário quebra as ligações C-N dando origem a um ácido carboxílico e uma amina primária (8). As regiões terminais dos monômeros, compostas por amino carboxi, estabilizam a estrutura dimérica da protease, dando a forma de folha β, entrelaçadas de forma antiparalelas, e por fim a protease apresenta duas abas que se movimentam para a entrada do substrato permitindo a interação enzima – substrato (15), conforme apresentado na Figura 4.
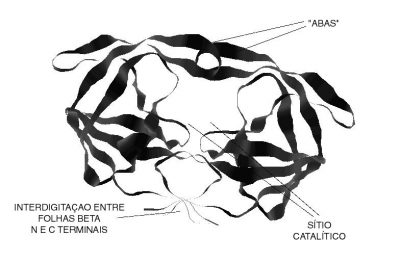
Figura 4: Enzima protease HIV (Peçanha et al, 2002)
Os inibidores da protease (IP) são substâncias químicas que possuem em sua estrutura uma ligação hidroxietileno ou hidroxietilamina que mimetizam o intermediário tetraédrico agindo de forma competitiva com o substrato natural, impedindo a clivagem das poliproteínas virais, interrompendo o ciclo do vírus HIV (8,17). A Figura 5 apresenta os fármacos IPs (20).
| Nome Genérico | Abreviatura | Nome Comercial | Fabricante | País de Origem |
| Amprenavir | APV | Agenerase® | GlaxoSmithKline | Inglaterra |
| Atazanavir | ATV | Reyataz® | Bristol-Myers Squibb | Estados Unidos |
| Darunavir | DRV | Prezista® | Tibotec, Inc. | Estados Unidos |
| Fosamprenavir | FPV | Lexiva® | GlaxoSmithKline | Inglaterra |
| Indinavir | IDV | Crixivan® | Merck | Alemanha |
| Lopinavir | LPV | Não informado | Não informado | Não informado |
| Lopinavir e Ritonavir | LPV/RTV | Kaletra® | Abbott Laboratories | Estados Unidos |
| Nelfinavir | NFV | Viracept® | Agouron Pharmaceuticals | Estados Unidos |
| Ritonavir | RTV | Norvir® | Abbott Laboratories | Estados Unidos |
| Saquinavir | SQV | Fortovase® | Hoffmann-La Roche | Suíça |
| Tipranavir | TPV | Aptivus® | Boehringer Ingelheim | Alemanha |
Figura 5: Fármacos Inibidores da Protease (adaptação Jota, 2011)
O Amprenavir é uma hidroxietil amino sulfonamida inibidor não-peptídico, aprovado em 1999 sendo ativo contra o HIV-1 e o HIV-2, atua através de sua ligação reversível com o local ativo da protease, com efeitos adversos sendo anormalidades gastrointestinal, hiperglicemia e parestesias (18,19).
Em 2003 o Atazanavir foi aprovado para utilização antirretroviral, ele é um azapeptídeo que é metabolizado quase que por completo através da isoenzima CYP3A4 do citocromo P450, seus metabólitos são eliminados pela bile na forma de metabólitos livre ou glucuronidado, sendo apenas 13% eliminados pela urina, desses 13% somente 7% na forma de medicamento inalterada. Seu efeito adverso característico é hiperbilirrubinemia pelo aumento da bilirrubina indireta, mas também pode causar náuseas e diarreia (18,19).
O Darunavir é um inibidor da dimerização e da atividade catalítica da protease, inibindo seletivamente a clivagem da poliproteína Gag – Pol, também é metabolizado pela isoenzima CYP3A4 do citocromo P450, seus metabólitos são eliminado 79,5% pelas fezes e 13,9% pela urina, é o antirretroviral da classe dos IP com a maior barreira genética ao desenvolvimento de resistência e costuma ser bem tolerada, com efeitos adversos diarreia, náuseas, infecção do trato respiratório superior, aumento da frequência urinária e batimentos cardíaco acelerado (18,19).
Fosamprenavir é um pró-fármaco que é hidrolisado rapidamente no epitélio intestinal, tornando-se o composto Amprenavir e fosfato inorgânico antes de atingir a circulação sistêmica. O Amprenavir como os anteriores é metabolizado pela a enzima CYP3A4 do citocromo P450, sendo inibidor do CYP3A4, pode aumentar as concentrações de drogas que são removidas por esta via metabólica, sendo excretado em menos de 1% de forma inalterada pela urina. Apresenta como efeitos adversos distúrbios gastrointestinais e hipertrigliceridemia (18,19).
O Indinavir é uma hidroxitilamina peptidomimética, que apresenta em sua estrutura química um inibidor de renina, responsável por impedir a clivagem de fenilalanina – prolina na poliproteína viral. Ele é formulado em forma de sal de sulfato para manter concentrações plasmáticas em níveis adequados após administração oral, sendo 10 vezes mais ativo contra o HIV-1 do que o HIV-2. Como efeitos adversos é característico os cristais medicamentosos na urina causando cólica renal, e em cerca de 3% dos pacientes apresentam nefrolitíase. Também pode ocorrer problemas como queda de cabelos, distúrbios gastrintestinais e sintomas do sistema nervoso central, como cefaleia e insônia (18,19).
Lopinavir / Ritonavir foram aprovados em 2000, juntos em uma combinação de 1:5 (Ritonavir: Lopinavir) aumentam os níveis plasmáticos do Lopinavir em concentrações viáveis para inibir a protease, ambos são metabolizados pela isoenzima CYP3A4 induzindo seu próprio metabolismo (CYP3A4), CYP2C9 e 2C19 e glucuronoconjugação. Sendo eliminados em menos de 3% por via renal de forma inalterado, 83% pelas fezes, e possui efeito adverso distúrbios gastrointestinais e elevações dos níveis de colesterol e triglicerídeos (18,19).
O Nelfinavir é um inibidor não-peptídico, que também é ativo contra o HIV-1 e o HIV-2, aprovado em 1997 apresentado em forma de sal mesilato de uma amina básica, seus efeitos colaterais comuns são a diarreia, diabetes, intolerância à glicose, e os níveis elevados de colesterol e triglicerídeos (18,19).
O Ritonavir é uma hidroxietilamina peptidomimética, aprovado em 1996 sendo mais ativo contra o HIV-1 do que contra o HIV- 2, é o primeiro inibidor da protease do HIV a trazer benefício na sobrevida dos pacientes. Seus efeitos adversos são gastrintestinais, dor abdominal, parestesias periféricas e periorais, e também provoca elevações de níveis de colesterol e triglicerídeos, aumentando o risco de aterosclerose a longo prazo de terapia (18,19).
O Saquinavir é um peptidomimético da classe hidroxietilamina com metabolismo extenso de primeira passagem ocorrendo através da enzima CYP3A4 citocromo P450 que é um inibidor muito fraco desta isoenzima, sendo eliminado por volta de 85% pelas fezes e 3% pela urina, sendo normalmente bem tolerada com efeitos adversos leves sendo distúrbios digestivos e metabólicas (18,19).
Tipranavir foi aprovado em 2005 sendo o único IP estruturalmente não peptideomimético, devido a isso lhe concedendo menor potência em relação aos demais, mas é indicado apenas em pacientes com falha terapêutica anterior. Ele contém em sua estrutura central um anel dihidropirona substituindo o núcleo peptidomimético hidroxietilênico, sendo metabolizado através do citocromo P450 3A4, e é um indutor de CYP2C9, CYP1A2, glucuronidação, glicoproteína-P e inibidor de CYP3A4 e 2D6. Apresenta um perfil de resistência com uma barreira genética mais elevada tendo os efeitos adversos como anormalidades gastrointestinal, aumento da taxa de triglicerídeos no sangue e disfunções metabólicas, bem como hepatotoxicidade (18,19). A figura a seguir representa a estrutura de todos os IPs.
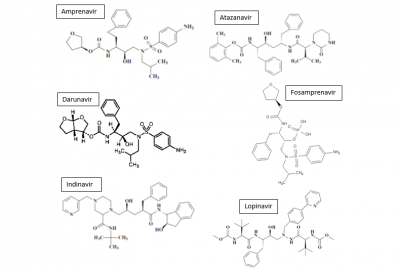

Figura 6: Estrutura química dos inibidores da protease (adaptado de Souza e Almeida, 2003)
Mutações da protease que conferem resistência aos Antirretrovirais
O HIV com genótipo alterado fica latente enquanto o selvagem é inibido pela TARV, e em poucas semanas o vírus que possui a resistência sai do estado de latência e se torna predominante, ocorrendo a falha terapêutica. Para correção do tratamento se introduz a terapia de resgate com um novo esquema de antirretrovirais, através do teste de genotipagem, onde se localiza a mutação do genoma viral que pode implicar a resistência aos fármacos e se determina um novo tratamento (21).
A Organização Mundial de Saúde (OMS) classifica como baixa (< 5%), intermediária (5 a 15%) e elevada (> 15%) a prevalência da transmissão do HIV resistente aos antirretrovirais, sendo que essa resistência vem crescendo e apresentando taxas elevadas nos últimos anos nos países com acesso à terapia anti-HIV. No Brasil, a prevalência tem sido considerada intermediária, com variações regionalizadas, como exemplo o que foi detectada na cidade de Santos-SP, sendo 36,8%, e em Salvador-BA de 18,9%. Portanto, a resistência transmitida está relacionada à localidade e quanto à manipulação antirretroviral, sendo difícil fazer generalizações muito amplas (22,23).
As mutações de resistência aos IPs levam a uma alteração na conformação tridimensional da protease, levando a uma diminuição do tempo de ligação entre os IPs e a enzima, e o tempo que a protease necessita para clivar seu substrato natural, tendo como resultado pela disputa ao sítio ativo a interação do substrato natural permitindo a clivagem das poliproteínas virais, e sua replicação. lém disso, um vírus necessita de um grupo de mutações para que possa equilibrar o seu fitness e replicar (24,25).
As mutações são apresentadas com abreviaturas de uma letra do aminoácido encontrado no vírus selvagem, seguido pela posição do mesmo e pela letra do aminoácido que representa a mutação, por exemplo a mutação na protease L90M, quer dizer que a leucina (L) na posição 90, foi substituída pela metionina (M), a Figura 7 apresenta as mutações da protease (18).
| W6R | R8K | R8Q | L10F | L10I | L10R | L10S | L10V | I11V | T12A |
| T12I | I13A | I13V | I15A | I15V | V15A | G16A | G16E | D17N | (G17N) |
| G17GR | Q18HL | Q18QL | Q18QI | K20I | K20M | K20R | K20T | A22AV | L23V |
| L24I | L24M | D25DH | A28S | D30N | L31LL | V32I | L33F | L33I | L33V |
| E34K | E34Q | E35D | E35EG | E35ETN | E35X | M36I | M36V | M36L | M36TNL |
| N37D | S37N | G40GK | R41K | R41T | K43T | E44P | K45I | K45R | M46F |
| M46I | M46L | I47A | I47L | I47V | G48E | G48V | G48M | I50L | I50V |
| G52S | F53L | I54L | I54T | I54V | I54M | K55R | K57R | R57K | Q58E |
| D60E | D60N | D60Y | Q61D | I62V | L63A | L63P | L63T | L63I | L63Q |
| L63V | I64V | H69N | H69Y | K70E | A71T | A71V | A71L | I72L | G73A |
| G73S | G73T | T74A | T74S | T74P | V75I | L76M | L76V | V77I | T80I |
| P81S | P81T | I82A | I82F | I82L | I82M | I82T | V82A | V82D | V82E |
| V82F | V82I | V82S | V82T | V82L | V82F | V82M | V82S | I84A | I84C |
| I84L | I84V | I85V | N88D | N88G | N88S | I89V | L89I | L89M | L89V |
| M89I | M89V | L90I | L90M | T91S | I93L | C95F | L97V | L99F |
Figura 7: Mutações de Resistência aos Inibidores da Protease (PIs)
(Adaptado de Stanford HIV Drug Resistance Database (http://hivdb.stanford.edu),2020)
As mutações no gene da protease que conferem resistência aos antirretrovirais é através das substituições nos aminoácidos localizados nas regiões essenciais à atividade da enzima, como no centro ativo, a estrutura flap e ou o local da ligação do substrato reduzindo a afinidade entre o inibidor e a protease mutante. E as mutações em outras regiões desestabilizam os dímeros alterando a conformação do centro ativo e da atividade enzimática, que podem ser compensadas através das mutações nos locais de clivagem Gag – Pol (17,18). A relação entre mutações da protease que conferem resistências aos IPs estão representadas na Figura 8 a seguir.
| IPs | Mutações Principais | Mutações acessórias |
| IDV | M46I/L,V82A/F/I/S/T,I84V/A/C | L10I/R/F/V, K20M/R/T/I, L24I, V32I, E35D, M36I/L/V, G48V, I54L/T/V, R57K, Q58E, L63A/I/P/Q/V/Y/T, A71T/V, G73S/T/C/A, L76V, V77I, N88D/S,L89M/V, L90M, I93L |
| RTV | V82A/F/I/S/T, I84V/A/C | L10I/R/F/V, G16E, K20M/R/T/I, L24I, V32I, L33I/F/V, E34K,M36I/L/V, G48V, F53L, I54L/T/V, Q58E, D60N, I62V, L63A/I/P/Q/V/Y/T, A71T/V, L90M |
| SQV | G48V, I84V/A/C, L90M | L10I/R/F/V, T12I, K20M/R/T/I, D30N, V32I, M36I/L/V, M46I/L,F53L, I54L/T/V, R57K, Q58E, D60N, I62V, L63A/I/P/Q/V/Y/T, A71T/V, G73S/T/C/A, T74S,L76M, V82A/F/I/S/T, N88D/S |
| NFV | D30N, L90M | L10I/R/F/V, I13V, K20M/R/T/I, M36I/L/V, M46I/L, G48V, I54L/T/V,Q58E, D60N, I62V, L63A/I/P/Q/V/Y/T, V77I, V82A/F/I/S/T, I84V/A/C, N88D/S, 93L |
| FPV/APV | I50V, I84V/A/C | L10I/R/F/V, L11I, 2K0M/R/T/I, 24I, V32I, L33I/F/V, R41K, K43R,M46I/L, I47A/V, G48M, I54L/T/V, 58E, L63A/I/P/Q/V/Y/T, 71V/T, G73S/T/C/A, L76V,V82A/F/I/S/T, L89V/T, L90M |
| LPV | Não possui | L10I/R/F/V, G16E, K20M/R/T/I, L24I, V32I, L33I/F/V, E34Q, M36I/L/V, K43T, M46I/L,I47A/V, G48V, I50V, F53L, I54L/T/V, Q58E, L63A/I/P/Q/V/Y/T, A71T/V, G73S/T/C/A, T74S,V82A/F/I/S/T, I84V/A/C, L89M/V, L90M, T91S |
| ATV | I50L, N88S, I84V/A/C | L10I/R/F/V, K20M/R/T/I, L24I, V32I, L33I/F/V, M36I/L/V, 45V,M46I/L, G48V, 53L, I54L/T/V, L63A/I/P/Q/V/Y/T, A71T/V, G73S/T/C/A, V82A/F/I/S/T,L88D/S/T, L89M/V, L90M |
| TPV | Não possui | L10I/R/F/V, 13V, I15V, K20M/R/T/I, 32I, L33I/F/V, E35D, M36I/L/V, N37D, R41K,K43T, K45I, M46L, I47A/V, I54L/T/V, 58E, D60N, A71T/V, 74P, V82T, 83D, I84V/A/C
|
| DRV | Não possui | L11L, I15V, V32I, L33F, E34V, 35G/N, 41I/T, I47F, I50V, I5454L/M/A/S/T/V, K70E, G73S, T74E/P, L76V, V82A/T/F/S/L/M, 84V/A/C, I85V, L89V, L90
|
Figura 8: Relação entre mutações da protease que conferem resistências aos IPs (Adaptado de Stanford HIV Drug Resistance Database (http://hivdb.stanford.edu),2020.)
Os IPs de modo geral apresentam a barreira genética para resistência as mutações maior que os demais classes dos ARVs, como exemplo, o tratamento contendo Ritonavir como complemento dos demais IPs, a protease viral necessita de múltiplas mutações para gerar resistência, sendo seu grau dependente do número e tipo de mutação presente. Normalmente as mutações acessórias se apresentam antes das principais, mas não apresentam um efeito significativo no fenótipo, pois servem para compensar o fitness replicativo, mas em conjunto com as mutações principais levam a uma resistência (9), conforme ilustrado na Figura 9 a seguir.
| IPs | Resistência Completa |
| APV/FPV | § 50V + 84V/A/C § 50V ou 84V/A/C + 2 de (10I/V/F/R, 11I, 20M/R/T/I, 24I, 32I,33F, 35D, 41K, 43R,46I/L,47V, 48M, 54L/V/M/A/T/S,58E,63A/I/P/Q/V/Y/T,71V/T,73S/T/C/A, 76V, 82A/F/I/T/S/M,89V/T, 90M) § 4 ou + de (10I/V/F/R, 11I, 20M/R/T/I, 24I, 32I, 33F, 35D, 41K, 43R, 46I/L, 47V, 48M, 54L/V/M/A/T/S, 58E, 63A/I/P/Q/V/Y/T, 71V/T, 73S/T/C/A, 76V, 82A/F/I/T/S/M, 89V/T, 90M) |
| APV/ RTV | § 8 ou + de (10I/V/F/R, 11I, 20M/R/T/I, 24I, 32I, 33F, 35D, 41K, 43R, 46I/L, 47V, 48M, 50V, 54L/V/M/A/T/S, 58E, 63A/I/P/Q/V/Y/T, 71V/T, 73S/T/C/A, 76V, 82A/F/I/T/S/M, 89V/T, 90M) |
| IDV | § 2 de (46I/L, 82A/F/I/S/T/M, 84V/A/C) § (46I/L ou 82A/F/I/S/T/M ou 84V/A/C) + 2 ou mais de (10I/V/R/F, 20M/R/T/I, 24I, 32I, 36I/L/V, 48V, 54L/T/V, 57K, 58E, 63A/I/P/Q/V/Y/T, 71T/V/I, 73S/T/C/A, 76V, 77I, 88D/S, 90M, 93L) § 4 ou + de (10I/V/R/F, 20M/R/T/I, 24I, 32I, 36I/L/V, 48V, 54L/T/V, 57K, 58E, 63A/I/P/Q/V/Y/T, 71T/V/I, 73S/T/C/A, 76V, 77I, 88D/S, 90M, 93L) |
| IDV/RTV | § 8 ou + de (10I/V/R/F, 20M/R/T/I, 24I, 32I, 36I/L/V, 46I/L, 48V,54L/T/V, 57K, 58E, 63A/I/P/Q/V/Y/T, 71T/V/I, 73S/T/C/A, 76V,77I, 82A/F/I/S/T/M, 84V/A/C, 88D/S, 90M, 93L) |
| RTV | § 82A/F/I/S/T/M e 84V/A/C § 82A/F/I/S/T ou 84V/A/C + pelo menos 2 (10I/R/V/F, 16E,20M/R/T/I, 24I, 32I, 33F, 36I/L/V, 46I/L, 48V, 53L, 54M/V/L,57K, 58E, 60N/E, 62V, 63P/A/I/Q/V/Y/T, 71V/T, 73S/T/C/A,90M) § 4 ou + de (10I/R/V/F, 16E, 20M/R/T/I, 24I, 32I, 33F, 36I/L/V,46I/L, 48V, 53L, 54M/V/L, 57K, 58E, 60N/E, 62V,63P/A/I/Q/V/Y/T, 71V/T, 73S/T/C/A, 90M) |
| SQV | § 2 de (48V, 84V/A/C ou 90M) § 1 de (48V, 84V/A/C ou 90M) + 2 de (10I/R/V/F, 20M/R/T/I,24I, 36I/L/V, 46I/L, 53L, 54L/M/V/T, 57K, 58E, 60N/E, 62V,63A/I/Q/V/Y/T/P, 71V/T/I, 73S/T/C/A, 74A/S, 82A/F/I/S/T/M,88D/S) § 4 ou + de (10I/R/V/F, 20M/R/T/I, 24I, 36I/L/V, 46I/L, 53L,54L/M/V/T, 57K, 58E, 60N/E, 62V, 63A/I/Q/V/Y/T/P, 71V/T/I,73S/T/C/A, 74A/S, 82A/F/I/S/T/M, 88D/S) |
| SQV/RTV | § 8 ou + de (10I/R/V/F, 20M/R/T/I, 24I, 36I/L/V, 46I/L, 53L,54L/M/V/T, 57K, 58E, 60N/E, 62V, 63A/I/Q/V/Y/T/P, 71V/T/I,73S/T/C/A, 74A/S, 82A/F/I/S/T/M, 88D/S) |
| LPV | § 8 ou mais de (10I/R/V/F, 16A/E, 20 I/M/R/T, 24I/V, 32I, 33F,34Q, 36I/V, 43T, 46I/L, 47 A/V, 48V, 50V, 53L, 54A/M//L/S/T/V,58E, 63 A/I/P/Q/V/Y/T, 71T/V/I, 73S/T/C/A/P, 74S,82A/F/I/S/T/M, 84A/C/V, 89M/V, 90M, 91S) |
| TPV/RTV | § 8 ou + de 10V/F/I, 13V, 20M/R/V, 32I, 33F, 35G, 36I, 43T, 45I,46L, 47V, 54A/M/V, 58E, 71V, 74P, 82L/T, 83D, 84V |
Figura 9: Relação do número de mutações da protease viral necessárias para gerar resistência completa aos IPs (Diaz, 2011)
Considerações Finais
Apresentamos 149 mutações de substituições, sendo 15 principais que estão associados a resistência aos IPs, com exceção ao Darunavir, Tipranavir e Lopinavir que perdem suas ações com um conjunto de mutações acessórias. As mutações ocorrem em 66 posições diferentes dos 99 aminoácidos da enzima protease, ou seja, os 33,3% restantes ao sofrerem substituição de aminoácidos não apresentam nenhum significado clínico e não interferem no fitness replicativo viral. Tem destaque a posição 82 que sofre 16 mutações diferentes como efetores de resistência em todos os IPs. O Saquinavir e o Indinavir são os dois inibidores de protease com a menor barreira genética de sua classe de ARV, pois necessitam de poucas substituições de aminoácidos para sofrerem uma resistência completa.
Os IPs são o grupo de ARV mais utilizados no mundo em combate a AIDS devido sua barreira genética elevada. De modo geral sofrem metabolização de primeira passagem através da enzima CYP3A4 citocromo P450, e apresentam como evento adverso mais comum o distúrbio gastrointestinal, ressaltando o Tipranavir que causa o efeito mais grave, a hepatoxicidade. Ele é o único que não é um peptidomimético, mas somente é usado quando os demais IPs sofreram resistência completa pelo vírus HIV.
Sabe-se que os portadores do HIV são imunocomprometidos, onde ao decorrer de suas vidas muitas doenças oportunistas são comuns a eles, devido a isso precisam de tratamento com fármacos que também sofrem na maioria das vezes metabolização pelo citocromo P450, fazendo assim uma interação entre os ARVs levando a uma anulação ou aumento de concentração no organismo, interferindo no tratamento antirretroviral.
Devido a enorme variabilidade genética do HIV que assim segue conferindo resistência ao tratamento, nota-se que o maior problema enfrentado está na forma de latência do mesmo, pois os ARVs são eficientes nas áreas que atacam o ciclo de vida. Sendo assim, destacam-se os IPs, que devido a sua maior barreira genética tornam as cargas virais indetectáveis, porém não ocorre a cura e os efeitos adversos tornam-se um problema para a adesão do tratamento.
Estima-se que no futuro exista a possibilidade de novos grupos de ARVs serem específicos ao DNA celular infectado com HIV, tendo como finalidade destruir o DNA viral latente, podendo gerar um grande efeito adverso, mas se for eficaz em trazer uma cura definitiva, os danos serão por um curto período de tempo, assim como a quimioterapia em tratamento de canceres, onde destrói as células cancerígenas, mas também destrói as células sadias, que em muitos casos traz a eficácia contra o câncer.
Também é possível serem criados ARVs que mimetizem os linfócitos T e macrófagos, com seus receptores de membrana (CD4) e coreceptores (CCR5 e CXCR4), para admitir a fusão do vírus, sem ter a maquinaria interna de uma célula comum para a replicação viral, encapsulando assim o vírus e o tornando inativo.
Referências
1– Baptista, V.I.C.S.P., Damaceno, D.S., Guarnieri, M.A.P., Junior, P.P., Souza, C.B. (2016). Fármacos de origem biotecnológica na terapêutica do vírus HIV. Revista UNILUS Ensino e Pesquisa, 13, 16-26.
2– UNAIDS. Estatísticas globais sobre HIV 2019; 2020 [acesso em 2020 Set29]. Disponível em: https://unaids.org.br/estatisticas/.
3– Ministério da saúde. Boletim epidemiológico HIV AIDS 2019; 2019 [acesso em 2020 Ago24]. Disponível em:http://www.aids.gov.br/pt-br/pub/2019/boletim-epidemiologico-hivaids-2019.
4– Carvalho, P.P., Barroso, S.M., Coelho, H.C., Penaforte, F.R.O.(2019). Fatores associados à adesão à terapia antirretroviral em adultos: revisão integrativa de literatura. Ciência e Saúde Coletiva, 24, 2543-2555.
5– Costa, N.R., Lago, R.F. (2010). Dilemas da política de distribuição de medicamentos antirretrovirais no Brasil. Ciência e Saúde Coletiva,15, 3529 – 3540.
6– Antunes, O.A.C., Peçanha, E.P., Tanuri, A. (2002). Estratégias farmacológicas para a terapia anti-aids. Química Nova, 25, 1108-1116.
7– Acurcio, F.A., Bonolo, P.F., Ceccato, M.G.B., Costa, J.O., Reis, E.A., Silveira, M.R.(2018). Efetividade da terapia antirretroviral na era de medicamentos em dose fixa combinada. Revista de Saúde Pública,52, 1-13.
8– Araújo, J.R.M., Ferreira, L.M., Freitas, C.A., Macêdo, O., Sousa, R.C.M., Vasconcelos, P.F.C.(2011). Genotipagem da resistência genotípica secundária aos antirretrovirais em pacientes com aids nos Estados do Pará e Amazonas, Brasil: 2002 a 2006. Revista Pan-Amazônica de Saúde, 2, 27-34.
9- Diaz, R.S. (2011). Guia para o manuseio de resistência antirretroviral. São Paulo. Permanyer Brasil Publicações.
10- Copeland, W.C., Day, B.J., Lewis W.(2003). Mitochondrial toxicity of NRTI antiviral drugs: an integrated cellular perspective. Nature Reviews Drug Discovery, 2, 812-822.
11– Grotto, R.M.T., Pardini, M.I.M.C.(2006). Biologia molecular do HIV-1 e genética da resistência humana à AIDS. Arquivos de Ciências da Saúde,13, 61-64.
12– Fink, H.T.K., Lima, L.M.P., Veloso, A.C.R. (2010). Resistência genotípica do vírus da imunodeficiência humana tipo 1 aos antirretrovirais. Comunicação em Ciências da Saúde, 21, 49-60.
13– Cunico, W., Gomes, C.R.B., Junior, W.T.V. (2008). HIV – recentes avanços na pesquisa de fármacos. Química Nova,31, 2111-2117.
14– Souza, M.V.N.(2005). Fuzeon, o primeiro medicamento de uma nova classe anti-HIV denominada inibidores de fusão. Revista Brasileira de Farmácia, 86, 112-116.
15-Almeida, M.V., Souza, M.V.N.(2003). Drogas anti-VIH: passado, presente e perspectiva futura. Química Nova, 26, 366-372.
16– Alvarenga, J.S.D., Moura, L.B., Nery, G.K.O., Silva, L.M.C.F.(2019). Prevalência de infecção pelo vírus da imunodeficiência humana (HIV) no município de Ariquemes – RO. South American Journal of Basic Education, Technical and Technological, 6, 256-267.
17– Fernandez, J.C.C., Fernandez, S.L.C., Gracie, R.S.G., Jesus, C.S., Rachid, M., Veloso, V.G., et al.(2005). Human immunodeficiency virus type 1 (HIV-1) genotyping in Rio de Janeiro, Brazil: assessing subtype and drug-resistance associated mutations in HIV-1 infected individuals failing highly active antiretroviral therapy. Memórias do Instituto Oswaldo Cruz,100, 73-78.
18- Costa, C.R. (2013). Manifestações bucais da AIDS e o perfil de mutações e de resistência do HIV em pacientes experimentando falha terapêutica. [Tese]. São Paulo: Faculdade de Odontologia da Universidade de São Paulo.
19– Jota, F.A. (2011). Os antirretrovirais através da história, da descoberta até os dias atuais. [Monografia]. Rio de Janeiro: Instituto de Tecnologia de Fármacos –Farmanguinhos/FIOCRUZ.
20– Alcantara, L.C.J., Araújo, A.F., Castro, B.G., Santos, E.S. (2009). Diversidade genética do vírus da imunodeficiência humana tipo 1 (HIV-1) em mulheres infectadas de uma cidade do nordeste do Brasil. Revista Brasileira de Ginecologia e Obstetrícia, 31, 609-614.
21-Andrade, C.H., Freitas, L.M., Oliveira, V. (2011). Twenty-Six Years of HIV science: an overview of anti-HIV drugs metabolism. Brazilian Journal of Pharmaceutical Sciences, 47, 209-230.
22– Brito, M.A. (2011). Fármacos recentes usados para o tratamento da infecção pelo HIV-1: enfuvirtida, maraviroc, raltegravir e etravirina. Revista de Ciências Farmacêutica Básica e Aplicada,32, 159-168.
23– Souza, M.V.N. (2005). Fármacos Inibidores de Fusão: uma Nova Estratégia no Combate à Replicação do Vírus VIH. Acta Farmacêutica Bonaerense,24, 291-299.
24- Azuka, C.O., Kenneth, A.A.(2013). Adverse drug reactions to antiretroviral therapy: Results from spontaneous reporting system in Nigeria. Perspectives in Clinical Research, 4, 117-124.
25-Eulálio, K.D., Luz, R.P. (2018). Resistência transmitida aos antirretrovirais em pacientes infectados pelo HIV-1 atendidos em um centro de referência. Jornal de Ciências da Saúde do Hospital Universitário da Universidade Federal do Piauí,1, 51-62.